

Table of Contents
Introduction: A Historic Milestone in Transplant Medicine
Hematopoietic stem cell transplantation (HSCT) is often a last, life‑saving option for patients with severe hematologic diseases. Yet even after a successful transplant, patients remain vulnerable to rare but devastating complications. Among the most feared is transplant‑associated thrombotic microangiopathy (TA‑TMA)—a condition historically linked with extremely high mortality and no FDA‑approved therapy.
That reality fundamentally changed with the Yartemlea FDA approval. In December 2025, the U.S. Food and Drug Administration approved Yartemlea (narsoplimab‑wuug), developed by Omeros Corporation, as the first and only treatment specifically indicated for TA‑TMA in adults and pediatric patients aged two years and older.
This approval is more than a regulatory milestone—it represents a paradigm shift in transplant medicine. For the first time, clinicians have access to a targeted, disease‑modifying therapy that addresses the underlying biology of TA‑TMA rather than relying solely on supportive care. The Yartemlea FDA approval redefines hope for transplant patients and their families.
Understanding Transplant‑Associated Thrombotic Microangiopathy (TA‑TMA)
TA‑TMA is a severe microvascular disorder that develops after HSCT due to widespread endothelial injury. Damage to the inner lining of small blood vessels triggers platelet activation and micro‑thrombus formation, leading to:
- Thrombocytopenia (low platelets)
- Hemolytic anemia
- Elevated lactate dehydrogenase (LDH)
- Progressive multi‑organ dysfunction
Organs most commonly affected include the kidneys, lungs, gastrointestinal tract, and central nervous system. Risk factors include conditioning regimens, calcineurin inhibitors, graft‑versus‑host disease (GVHD), and infections.
Historically, mortality rates exceeded 50% in high‑risk patients. With no approved therapy, management was limited to removing triggering agents and providing supportive care—often too late to stop disease progression. The Yartemlea FDA approval directly addresses this unmet medical need.
The Science Behind Yartemlea: Mechanism of Action (MOA)
The foundation of the Yartemlea FDA approval lies in its highly selective mechanism of action.
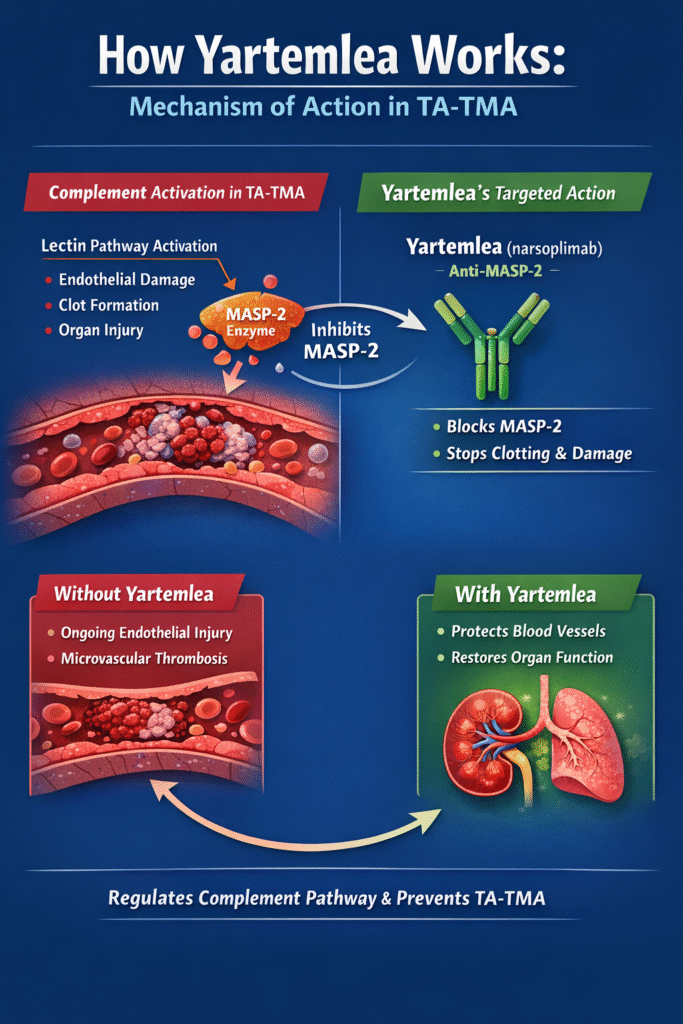
Yartemlea (narsoplimab‑wuug) is a fully human monoclonal antibody that inhibits mannan‑binding lectin‑associated serine protease‑2 (MASP‑2), the key effector enzyme of the lectin pathway of the complement system.
Why MASP‑2 Matters in TA‑TMA
- The complement system has three pathways: classical, alternative, and lectin
- In TA‑TMA, the lectin pathway becomes pathologically overactivated
- MASP‑2 activation leads to endothelial damage, inflammation, and microvascular thrombosis
By selectively blocking MASP‑2, Yartemlea:
- Halts lectin pathway–mediated endothelial injury
- Reduces micro‑thrombus formation
- Preserves the classical and and alternative complement pathways
This precision allows immune defense against infections to remain largely intact—an essential benefit for immunocompromised transplant patients. This targeted biology was a key factor behind the success of the Yartemlea FDA approval.
Detailed FDA Approval Information
The Yartemlea FDA approval was granted on December 24, 2025, for the treatment of TA‑TMA in adults and pediatric patients aged ≥2 years.
Special FDA Designations
Due to the severity and rarity of TA‑TMA, Yartemlea received multiple expedited designations:
- Breakthrough Therapy Designation – Recognizing substantial improvement over existing care
- Orphan Drug Designation – For treatment of a rare disease
- Priority Review – Accelerated FDA evaluation timeline
Approval was based on a single‑arm, open‑label clinical trial supported by data from an Expanded Access Program (EAP)—a regulatory pathway often used when randomized trials are not feasible for life‑threatening rare diseases.
Clinical Success: Evidence Supporting the Yartemlea FDA Approval
Key Efficacy Outcomes
The pivotal TA‑TMA study enrolled 28 patients, with an additional 19 patients treated through the EAP.
- TMA Response Rate: 61% in the pivotal study
- Expanded Access Response: 67% (pediatric), 69% (adult)
- 100‑Day Survival Rate: 73–74% from TA‑TMA diagnosis
These outcomes represent a dramatic improvement compared with historical survival data and were central to securing the Yartemlea FDA approval.
Safety Profile and Side Effects
Importantly, the Yartemlea FDA approval does not include a Boxed Warning or REMS program, simplifying access and clinical use.
Common Adverse Effects (≥20%)
- Hemorrhage
- Diarrhea
- Serious infections
- Nausea and vomiting
- Neutropenia
- Fever (pyrexia)
- Fatigue
- Hypokalemia
Serious Risks
- Life‑threatening infections occurred in ~36% of patients
- Includes sepsis, pneumonia, viral infections, and bacteremia
Patients receiving Yartemlea require close monitoring, particularly for infectious complications. Despite these risks, the FDA concluded that the benefit‑risk balance strongly favored approval.
Drug Interactions: A Practical Advantage
As a monoclonal antibody, Yartemlea is not metabolized by CYP450 enzymes, which significantly reduces the risk of pharmacokinetic drug interactions.
Clinical Implications
- Compatible with complex transplant drug regimens
- Minimal interaction with immunosuppressants and antimicrobials
- Easier integration into standard HSCT protocols
This favorable interaction profile further enhances the real‑world value of the Yartemlea FDA approval.
Conclusion: Redefining Hope in TA‑TMA
The Yartemlea FDA approval marks a turning point in transplant medicine. For the first time, clinicians can directly target the biological driver of TA‑TMA rather than relying on supportive care alone.
With its selective complement inhibition, strong survival data, and manageable safety profile, Yartemlea establishes a new standard of care for a previously untreatable condition. Most importantly, it restores hope to transplant patients facing one of the most devastating post‑transplant complications.